Example: Making Morpheus Peptagrams
Morpheus is a fast search engine designed for high-quality data. As Morpheus does not come with a bundled viewer, peptagram provides a unique tool to view Morpheus search results.
The scripts that process Morpheus have morpheus_peptagram in their name:
mac_morpheus_peptagram.command- which can be clicked in Finder on Mac OSXwin_morpheus_peptagram.bat- which can be clicked in WIndows Filer Explorerdo_morpheus_peptagram.py- which is run on the command-line aspython do_morpheus_peptagram.py -i
Test
To run an automated test, unzip example_data.zip. It should create an example_data directory containing sub-directories such as morpheus, mascot etc. Then to run the test:
python do_morpheus_peptagram.py test
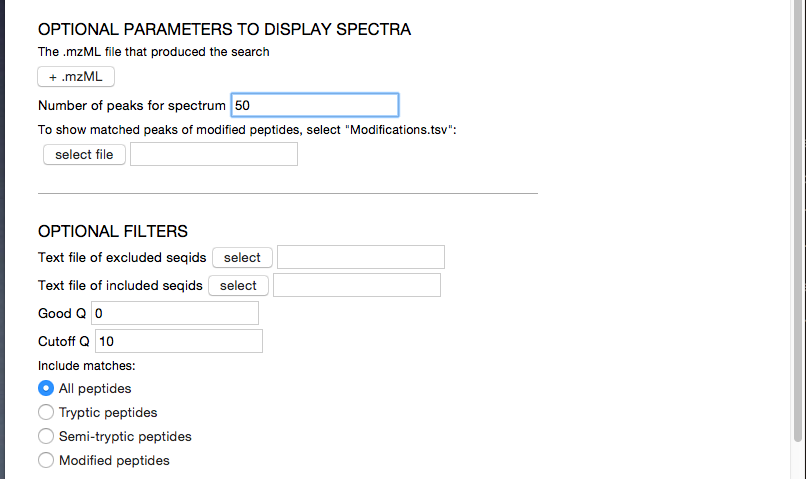
Create a Peptagram with the GUI
To use morpheus_peptagram, first start the program, then you should get a window that looks something like this:
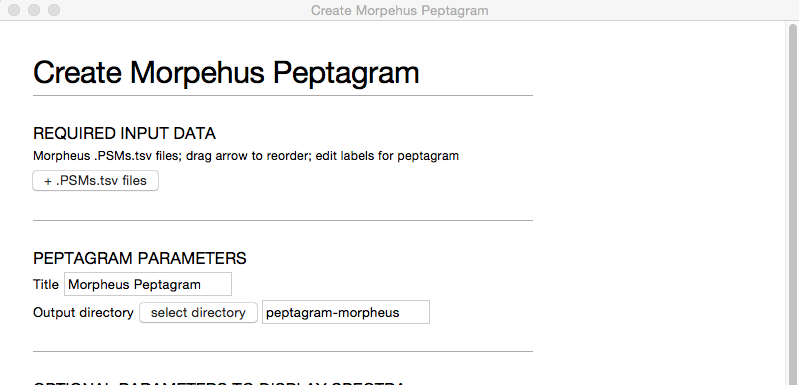
To load the Morpheus files, click + PSMs.tsv files and select all the .PSMs.tsv files that you want to compare. morpheus_peptagram will figure out the corresponding .protein_group.tsv files from these filenames.
Once selected, you'll see a list of files:

You can now reorder the .PSMs.tsv files into your preferred order by dragging the ☰ icon.
Then you can scroll down to the bottom, and click the submit button:

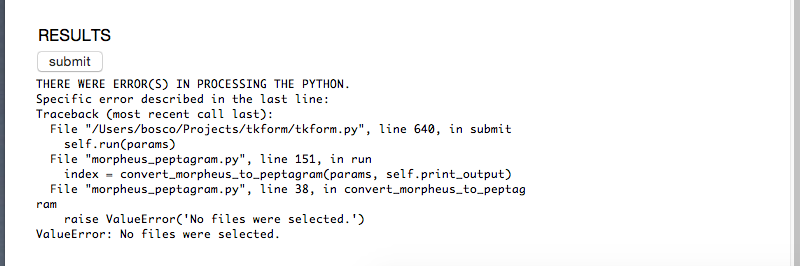
If there are any errors encountered, they'll appear below the submit button. Hopefully the error message at the last line will help you trouble-shoot the problem, and then you can click submit again.
If it worked, you'll get a link to a newly created directory containing your peptagram:
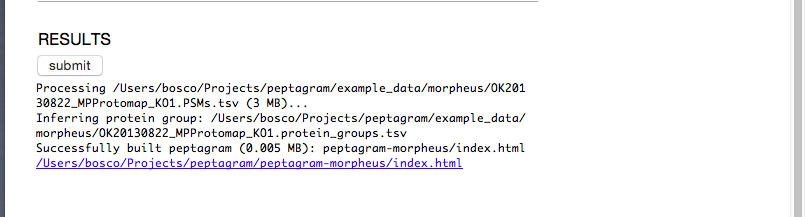
Later, you might want to tweak the options, such as loading the spectra from the original .mzML files, or restricting the display to matches by the Q-score.